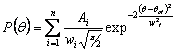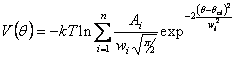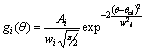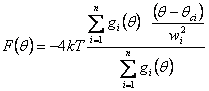|
 |
 |
|
|
Research at the MoLNaC is geared towards the interface between
catalysis and material science.
Understanding (and possibly solving) chemical problems, especially of
industrial relevance, is our main activity. To this end we use the
armory of tools known as computational chemistry. The areas of
interest span from understanding structure and function relationship
in organometallic compounds, to unraveling the mechanistic of
catalysts at work, to soft condensed matter simulations. Finally, we
are not shy to tackle systems of biological interest. In all cases we
try to interact as much as possible with experimentalists. Often we
are unsatisfied with the available tools and/or models and we develop
new ones. Particular interest is devoted to development of methods for
modelling systems across time and length scales.
Hybrid Quantum Mechanical Molecular
Mechanics Methods. In collaboration with the group of Tom
Ziegler, we have been deeply involved in the development of hybrid QM/MM
methods within the ADF package. QM/MM approaches are of remarkable relevance
since they allow to work on real size systems. That is, no reduction of the
system to an oversimplified and chemically poorly relevant model is needed Dispersion interaction within DFT. We are working to implement a modified
version of Grimmes approach to include dispersion interaction within DFT. Calculation of solvent accessible surface
areas. In collaboration with a bioinformatics
group we developed a tool for the fast calculation of solvent accessible
surface areas of proteins and nucleic acids, POPS, that works at atomic level
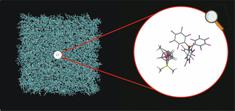
and at coarse grained level. Ion-pairing in Organometallics. Recently, we have developed a protocol,
based on a classical Molecular Dynamics approach, to study organometallic ion
pairs in low polarity solvents, see Figure. Schematic
representation of the system used to simulate a metallocenium ion-pair in
benzene solution. Force-Fields for common organic solvents:
Benzene and Cyclohexane.
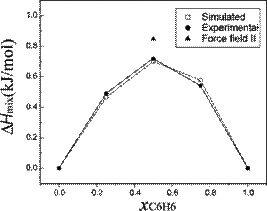
We investigated mixtures of cyclohexane
and benzene by means of all-atom molecular dynamics simulations. The force
fields have been validated calculating thermodynamic properties such as density
and enthalpy of mixing and diffusion coefficient for five different
compositions of benzene/cyclohexane mixtures and compared with experimental
results. Calculated and
experimental enthalpies of mixing (kJ/mol). The value of enthalpy of mixing
obtained using a different force field for the 1:1 mixture is also reported.
References
Polymer Coarse-Graining.
High molecular weight polymer chains are
difficult to relax. The longest relaxation of an entangled polymer melt of
length N scales at least as N3, giving at least N4
in cpu time, this way is only feasible for relatively short chain lengths. One
way to circumvent this problem is to reduce the degrees of freedom by
coarsening the models and keeping only those degrees of freedom that are deemed
relevant for the particular range of interest.
Recently, in collaboration with Florian
Mueller-Plathe (TU-Darmstadt, Germany) we developed new tools and models in the
framework of systematic polymer coarse-graining.
The main point of this class of methods is the
mapping of atomistic features to mesoscopic models. To this end, one groups
several atom together into super-atoms.
Mesoscale Potentials:
we introduced a new
class of mesoscale potentials based on sum of
Gaussian functions. A multipeaked distribution of a structural parameter q can
be approximated by a sum of n Gaussian functions characterized by their centres (qci),
total area (Ai) and width (wi): Given
a distribution P(q) of
some structural parameter q such as a bond length or an angle, a first
approximation of the corresponding potential can be derived doing a simple
Boltzmann inversion of P(q). The corresponding potential obtained by
Boltzmann inversion can be written as: If we define For the bond potentials it
is possible to derive equations similar to eq. 3 and eq. 4 for potential and
forces, respectively.
References
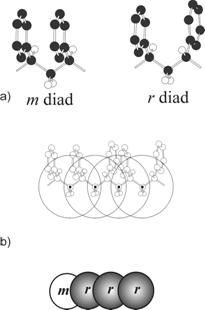
Realistic
Models of Vinyl Polymers: we introduced a
systematic procedure to coarse-grain atomistic models of the largest family of
synthetic polymers into a mesoscopic model that is able to keep detailed
information about chain stereosequences. The mesoscopic model consists of
sequences of superatoms centered on methylene carbons of two different types
according to the kind of diad (m or r) they belong to. The
proposed mesoscale model has been successfully tested against structural and dynamical
properties for different chain lengths and opens the possibility of relaxing
melts of high molecular weight vinyl polymers. a) Polystyrene m and r diads in
transplanar conformation (hydrogen atoms on phenyl rings are omitted for
clarity). b) Illustration of the mapping scheme for polystyrene:one bead
corresponds to a diadic m or r unit. The center of these super-atoms, as indicated
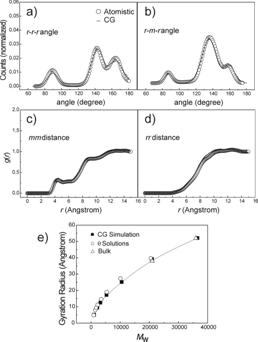
by filled squares, are the methylene carbons. Histograms (at 500 K) of a) r-r-r;
b) r-m-r ;angle extracted from atomistic (empty circles)
and from coarse-grained simulations (solid lines). Interchain c) m-m ;
d) r-r pair correlation function from atomistic simulations (empty
circles) and obtained with the coarse grained model (solid lines). e)Values of
gyration radii
References
Reverse-Mapping:
systematic procedures to obtain well relaxed atomistic melt structures from
mesocale models are important tools to obtain reliable and detailed models of
polymers in bulk. A fast and efficient reverse-mapping
procedure, successfully tested against experimental data, allows to obtain
atomistic models of both stereoregular and stereoirregular polymers and opens
the possibility of relaxing large molecular weight melts of vinyl chains. The mesocale and the reconstructed atomistic
models of a 350-mer chains of atactic polystyrene. The read beads are the meso
and the yellow ones the racemo superatoms.
References
Automatic Optimization of Force-Fields. Fast generation of accurate force fields
without the frustrations of parameterisation by hand. We developed a fast and
accurate method and the relative software for automatic optimization of point
charges from ab-initio calculations. |
||