|
 |
 |
|
|
Research at the MoLNaC is geared towards the interface between
catalysis and material science.
Understanding (and possibly solving) chemical problems, especially of
industrial relevance, is our main activity. To this end we use the
armory of tools known as computational chemistry. The areas of
interest span from understanding structure and function relationship
in organometallic compounds, to unraveling the mechanistic of
catalysts at work, to soft condensed matter simulations. Finally, we
are not shy to tackle systems of biological interest. In all cases we
try to interact as much as possible with experimentalists. Often we
are unsatisfied with the available tools and/or models and we develop
new ones. Particular interest is devoted to development of methods for
modelling systems across time and length scales.
Quantum Mechanical Studies. We have been happy that bioinformatics
groups asked for our QM expertise. Of course, they provided all the biological
background and expertise. This way, we contributed to characterize rhodamine, a
fluorescent probe used to follow dynamics of proteins. Recently, we started to
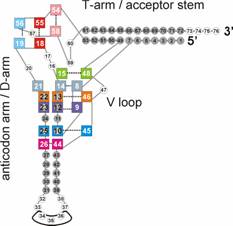
work on H-bonded nucleic base complexes, and we contributed to the first
characterization of the full set of the nine tertiary interactions in tRNA, see
Figure. Particular attention has been focused on the effect of chemical
modifications and of metal binding. Classical and
Steered Molecular Dynamics Studies. Interaction of Synthetic Polymers and Biomembranes. The
understanding of interactions of polyethylene glycol (PEG) or polyethylenoxide
(PEO) with biological interfaces has important technological application in
industry and in medicine. In collaboration with Danilo Roccatano (International University Bremen, Germany) and Sandeep Pal (International Centre for Life Newcastle, UK) we studied
structural and dynamical
properties of the PEO at dimyristoylphospatidylcholine (DMPC) bilayer/water
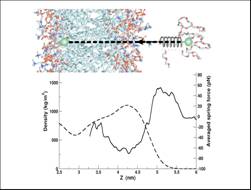
interface by Molecular Dynamics (MD) and Steered Molecular Dynamics (SMD) DMPC
density profile (dotted line) and the running averaged force from pulling experiments
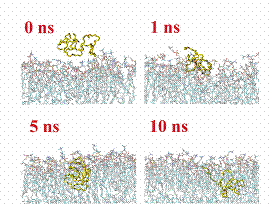
(bold line) versus Z distance of the PEO centre of mass. The insets on right
show snapshots of the trajectory along one of the pulling experiments. PEO chain
is in yellow.
References
|
||